What's new in the RDKit since the 2013 UGM?¶
General¶
- The RDKit now supports both python3 and python2. More from Riccardo Vianello later.
- There is now conda (https://github.com/conda/conda) support for the RDKit. This integrates the RDKit with the anaconda python distribution (http://docs.continuum.io/anaconda/index.html) and makes distributing binaries easier. More from Riccardo Vianello later.
- The github repo is linked to the Travis CI system; all commits are automatically built and tested. Original contribution from Maciej Wójcikowski
Contrib Area¶
- Fraggle similarity algorithm implementation from Jameed Hussain (https://github.com/rdkit/UGM_2013/blob/master/Presentations/Hussain.Fraggle.pdf)
- Code for building models from ChEMBL from Paul Czodrowski. Corresponds to the paper: Czodrowski, P. (2013). hERG me out. Journal of chemical information and modeling, 53(9), 2240-2251.
- Conformer parser from Sereina Riniker. More on this later.
New Feature Highlights¶
- Richer QueryAtom functionality
- ForceField constraints (from Paolo Tosco)
- PandasTools improvements (from Samo Turk)
- FMCS implementation in C++ (from Alexander Savelyev)
- SimpleEnum functionality
- 3D structure viewing in IPython notebook (from Patrick Fuller)
- Chemical reaction improvements (from Nadine Schneider)
Demos¶
import gzip
from rdkit import Chem
from rdkit.Chem import Draw,AllChem
from rdkit.Chem.Draw import IPythonConsole
New QueryAtom functionality¶
The idea here is to make it a lot easier and faster to find all the atoms in a molecule that match a particular query.
from rdkit.Chem import rdqueries
inf = gzip.open('./data/zim.sdf.gz','r')
zim_mols = [x for x in Chem.ForwardSDMolSupplier(inf) if x is not None]
len(zim_mols)
11004
%timeit [len([x for x in y.GetAtoms() if x.GetHybridization() == Chem.HybridizationType.SP2]) for y in zim_mols]
1 loops, best of 3: 1.06 s per loop
v1=[len([x for x in y.GetAtoms() if x.GetHybridization() == Chem.HybridizationType.SP2]) for y in zim_mols]
qa = rdqueries.HybridizationEqualsQueryAtom(Chem.HybridizationType.SP2)
v2=[len(y.GetAtomsMatchingQuery(qa)) for y in zim_mols]
v1==v2
True
qa = rdqueries.HybridizationEqualsQueryAtom(Chem.HybridizationType.SP2)
%timeit [len(y.GetAtomsMatchingQuery(qa)) for y in zim_mols]
10 loops, best of 3: 104 ms per loop
--------------------------------------------------------------------------- AttributeError Traceback (most recent call last) <ipython-input-15-c819d19e2eda> in <module>() ----> 1 qa.GetQuery().SetNegation(True) AttributeError: 'QueryAtom' object has no attribute 'GetQuery'
dir(rdqueries)
['AtomNumEqualsQueryAtom', 'AtomNumGreaterQueryAtom', 'AtomNumLessQueryAtom', 'ExplicitDegreeEqualsQueryAtom', 'ExplicitDegreeGreaterQueryAtom', 'ExplicitDegreeLessQueryAtom', 'ExplicitValenceEqualsQueryAtom', 'ExplicitValenceGreaterQueryAtom', 'ExplicitValenceLessQueryAtom', 'FormalChargeEqualsQueryAtom', 'FormalChargeGreaterQueryAtom', 'FormalChargeLessQueryAtom', 'HCountEqualsQueryAtom', 'HCountGreaterQueryAtom', 'HCountLessQueryAtom', 'HybridizationEqualsQueryAtom', 'HybridizationGreaterQueryAtom', 'HybridizationLessQueryAtom', 'InNRingsEqualsQueryAtom', 'InNRingsGreaterQueryAtom', 'InNRingsLessQueryAtom', 'IsAliphaticQueryAtom', 'IsAromaticQueryAtom', 'IsInRingQueryAtom', 'IsUnsaturatedQueryAtom', 'IsotopeEqualsQueryAtom', 'IsotopeGreaterQueryAtom', 'IsotopeLessQueryAtom', 'MassEqualsQueryAtom', 'MassGreaterQueryAtom', 'MassLessQueryAtom', 'MinRingSizeEqualsQueryAtom', 'MinRingSizeGreaterQueryAtom', 'MinRingSizeLessQueryAtom', 'RingBondCountEqualsQueryAtom', 'RingBondCountGreaterQueryAtom', 'RingBondCountLessQueryAtom', 'TotalDegreeEqualsQueryAtom', 'TotalDegreeGreaterQueryAtom', 'TotalDegreeLessQueryAtom', 'TotalValenceEqualsQueryAtom', 'TotalValenceGreaterQueryAtom', 'TotalValenceLessQueryAtom', '__doc__', '__file__', '__name__', '__package__']
ForceField constraints¶
From Paolo's announcement:
- support for fixed atoms was already there, but could be accessed only
from C++. I exposed it in Python through the AddFixedPoint() method
- Python support for distance constraints was already there through the
AddDistanceConstraint() method. However, now there are two more functions, namely UFFAddDistanceConstraint() and MMFFAddDistanceConstraint(), which compared to AddDistanceConstraint() accept one more boolean parameter (i.e., relative). If relative is True, then minLen and maxLen values are intended as deltas relative to the current value
- Similarly, now there are UFFAddAngleConstraint(),
MMFFAddAngleConstraint(), UFFAddTorsionConstraint(), MMFFAddTorsionConstraint() methods to constrain angles and dihedrals
- To harmonically restrain individual atoms to their original positions,
there are UFFAddPositionConstraint() and MMFFAddPositionConstraint() All constraints are implemented as flat-bottomed potentials, which are effective only when the target property is below min (distance, angle and dihedral constraints) or above max (distance, angle, dihedral and position constraints).
One usage example: http://rdkit.blogspot.ch/2013/12/using-allchemconstrainedembed.html
PandasTools Improvements¶
import pandas as pd # Import pandas
from rdkit.Chem import PandasTools
inf = gzip.open('./data/zim.sdf.gz','r')
cpds = PandasTools.LoadSDF(inf, includeFingerprints=True)
cpds.columns
Index([u'ID', u'SMILES', u'ROMol'], dtype='object')
from rdkit.Chem import Descriptors
cpds['logp'] = cpds['ROMol'].map(Descriptors.MolLogP)
cpds['mw'] = cpds['ROMol'].map(Descriptors.MolWt)
PandasTools.FrameToGridImage(cpds.head(8), legendsCol="ID", molsPerRow=4)

# pull out everything matching a carbazole scaffold:
qry=Chem.MolFromSmiles('N1C2=CC=CC=C2C2=C1C=CC=C2')
filtered = cpds[cpds['ROMol']>=qry]
len(filtered)
23
qry=Chem.MolFromSmarts('c1ncc[n,c]c1')
filtered2 = cpds[cpds['ROMol']>=qry]
len(filtered2)
--------------------------------------------------------------------------- ValueError Traceback (most recent call last) <ipython-input-18-d5475fa45b2d> in <module>() 1 qry=Chem.MolFromSmarts('c1ncc[n,c]c1') ----> 2 filtered2 = cpds[cpds['ROMol']>=qry] 3 len(filtered2) 4 /Library/Python/2.7/site-packages/pandas/core/ops.pyc in wrapper(self, other) 570 571 # scalars --> 572 res = na_op(values, other) 573 if np.isscalar(res): 574 raise TypeError('Could not compare %s type with Series' /Library/Python/2.7/site-packages/pandas/core/ops.pyc in na_op(x, y) 533 result = lib.vec_compare(x, y, op) 534 else: --> 535 result = lib.scalar_compare(x, y, op) 536 else: 537 /Library/Python/2.7/site-packages/pandas/lib.so in pandas.lib.scalar_compare (pandas/lib.c:12114)() /Users/landrgr1/RDKit_git/rdkit/Chem/PandasTools.pyc in _molge(x, y) 153 if hasattr(x,'_substructfp'): 154 if not hasattr(y,'_substructfp'): --> 155 y._substructfp=_fingerprinter(y,True) 156 if not DataStructs.AllProbeBitsMatch(y._substructfp,x._substructfp): 157 return False /Users/landrgr1/RDKit_git/rdkit/Chem/PandasTools.pyc in <lambda>(x, y) 141 try: 142 from rdkit.Avalon import pyAvalonTools as pyAvalonTools --> 143 _fingerprinter=lambda x,y:pyAvalonTools.GetAvalonFP(x,isQuery=y,bitFlags=pyAvalonTools.avalonSSSBits) 144 except ImportError: 145 _fingerprinter=lambda x,y:Chem.PatternFingerprint(x,fpSize=2048) ValueError: Sanitization error: Can't kekulize mol
PandasTools.FrameToGridImage(filtered.head(8), legendsCol="ID", molsPerRow=4)

from rdkit.Chem import AllChem
AllChem.Compute2DCoords(qry)
for mol in filtered['ROMol']:
AllChem.GenerateDepictionMatching2DStructure(mol,qry)
PandasTools.FrameToGridImage(filtered.head(8), legendsCol="ID", molsPerRow=4)

FMCS in C++¶
Rewrite in C++ of Andrew Dalke's FMCS algorithm. It includes some small API changes and algorithmic tweaks.
The new code is already documented in the Getting Started book.
Here's an example showing one of the interesting new features of the code that can help identify scaffolds that have some variation within the scaffold itself:
from rdkit.Chem import rdFMCS
with open('data/Target_no_130_60894.txt') as inf:
ms = [Chem.MolFromSmiles(x.strip().split()[-1]) for x in inf]
ms1 = [m for m in ms if m is not None]
with open('data/Target_no_121_20096.txt') as inf:
ms = [Chem.MolFromSmiles(x.strip().split()[-1]) for x in inf]
ms2 = [m for m in ms if m is not None]
mcs= rdFMCS.FindMCS(ms1,completeRingsOnly=True,timeout=60)
print mcs.smartsString
[#6]1:[#6]:[#6]:[#6]:[#6]:[#6]:1
mcs= rdFMCS.FindMCS(ms1,
atomCompare=rdFMCS.AtomCompare.CompareAny,
bondCompare=rdFMCS.BondCompare.CompareAny,
completeRingsOnly=True,timeout=60)
print mcs.smartsString
[#6]1:[#6]:[#6]:[#6](:[#6]:[#6]:1)-[#8,#6,#16]-[#6]-[#6]-[#6]-[#7]1-[#6]-[#6]-[#7,#6](-[#6]-[#6]-1)-[#6]1:[#6,#7]:[#6]:[#6]:[#6]:[#6,#7]:1
As a reminder of what the previous code generates:
from rdkit.Chem import MCS
mcs= MCS.FindMCS(ms1,atomCompare='any',bondCompare='any',completeRingsOnly=True)
print mcs.smarts
[*]~@1~@[*]~@[*]~@[*](~@[*]~@[*]~@1)~!@[*]~!@[*]~!@[*]~!@[*]~!@[*]~@1~@[*]~@[*]~@[*](~@[*]~@[*]~@1)~!@[*]~@1~@[*]~@[*]~@[*]~@[*]~@[*]~@1
Here's another example:
mcs= rdFMCS.FindMCS(ms2,completeRingsOnly=True,timeout=60)
print mcs.smartsString
[#6]1-[#6]-,:[#6]-[#6](-[#6]-[#6]-1)-[#7]
mcs= rdFMCS.FindMCS(ms2,
atomCompare=rdFMCS.AtomCompare.CompareAny,
bondCompare=rdFMCS.BondCompare.CompareAny,
completeRingsOnly=True,timeout=60)
print mcs.smartsString
[#6]1-[#6]-,:[#6]-[#6](-[#6]-[#6]-1)-[#7]1-[#6]-[#6]-[#6,#7](-[#6]-[#6]-1)-[#6]-[#6]1:[#6]:[#6]:[#6]:[#6]:[#6]:1-[#17,#6,#8,#9,#35,#53]
It would be nice to show a rendering of the scaffold here, but the current depiction code doesn't do a particularly good job with list-queries.
We can, of course, use these scaffolds to get nice aligned depictions:
import copy
core = Chem.MolFromSmarts(mcs.smartsString)
AllChem.Compute2DCoords(core)
mscp = copy.deepcopy(ms2)
[AllChem.GenerateDepictionMatching2DStructure(x,core) for x in mscp]
Draw.MolsToGridImage(mscp,molsPerRow=4)

SimpleEnum¶
This brings into the RDKit core some convenience functionality that I've previously talked about.
Here's the reaction we're working with:
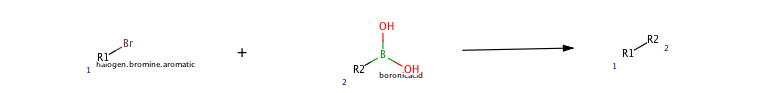
rxnB="""$RXN
ISIS 052820091627
2 1
$MOL
-ISIS- 05280916272D
2 1 0 0 0 0 0 0 0 0999 V2000
-3.2730 -7.0542 0.0000 Br 0 0 0 0 0 0 0 0 0 0 0 0
-3.9875 -7.4667 0.0000 R# 0 0 0 0 0 0 0 0 0 1 0 0
1 2 1 0 0 0 0
V 1 halogen.bromine.aromatic
M RGP 1 2 1
M END
$MOL
-ISIS- 05280916272D
4 3 0 0 0 0 0 0 0 0999 V2000
3.4375 -7.7917 0.0000 R# 0 0 0 0 0 0 0 0 0 2 0 0
4.1520 -7.3792 0.0000 B 0 0 0 0 0 0 0 0 0 0 0 0
4.1520 -6.5542 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
4.8664 -7.7917 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2 3 1 0 0 0 0
1 2 1 0 0 0 0
2 4 1 0 0 0 0
V 2 boronicacid
M RGP 1 1 2
M END
$MOL
-ISIS- 05280916272D
2 1 0 0 0 0 0 0 0 0999 V2000
11.2667 -7.3417 0.0000 R# 0 0 0 0 0 0 0 0 0 1 0 0
11.9811 -6.9292 0.0000 R# 0 0 0 0 0 0 0 0 0 2 0 0
1 2 1 0 0 0 0
M RGP 2 1 1 2 2
M END
"""
rxn = AllChem.ReactionFromRxnBlock(rxnB)
rxn.Initialize()
r1 = rxn.GetReactantTemplate(0)
for smi in ('CCBr','c1ccccc1Br'):
tm=Chem.MolFromSmiles(smi)
print(smi,tm.HasSubstructMatch(r1),rxn.IsMoleculeReactant(tm))
('CCBr', True, True)
('c1ccccc1Br', True, True)
from rdkit.Chem.SimpleEnum import Enumerator
nWarn,nError,nReacts,nProds,reactantLabels = Enumerator.PreprocessReaction(rxn)
print(reactantLabels)
(((0, 'halogen.bromine.aromatic'),), ((1, 'boronicacid'),))
for smi in ('CCBr','c1ccccc1Br'):
tm=Chem.MolFromSmiles(smi)
print(smi,tm.HasSubstructMatch(r1),rxn.IsMoleculeReactant(tm))
('CCBr', False, False)
('c1ccccc1Br', True, True)
3D molecules in IPython notebook¶
cpds['SMILES'][2]
'OC(c1ccncc1)c1ccc(OCC[NH+]2CCCC2)cc1'
m1 = Chem.MolFromSmiles('OC(c1ccncc1)c1ccc(OCC[NH+]2CCCC2)cc1')
m1

from rdkit.Chem import AllChem
m1h = Chem.AddHs(m1)
AllChem.EmbedMolecule(m1h)
AllChem.MMFFOptimizeMolecule(m1h)
m1h

IPythonConsole.ipython_3d=True
m1h
m1 = Chem.MolFromPDBFile('./data/3E4.pdb')
m1=AllChem.AssignBondOrdersFromTemplate(\
Chem.MolFromSmiles('Cc1cc(c2ccccc2c1Oc3c(cccn3)c4ccnc(n4)NC5CCC(CC5)N)NS(=O)(=O)c6ccccc6Cl'),
m1)
m1
m2 = Chem.MolFromPDBFile('./data/3EL.pdb')
m2=AllChem.AssignBondOrdersFromTemplate(\
Chem.MolFromSmiles('c1ccc(cc1)S(=O)(=O)Nc2ccc(c3c2cccc3)Oc4c(cccn4)c5ccnc(n5)NC6CCC(CC6)N'),
m2)
m2
o3=AllChem.GetO3A(m2,m1)
print o3.Align()
print o3.Score()
0.405595407951 231.074602218
Chem.CombineMols(m1,m2)